Specificity in the response to growth factors: TGFß regulation.
TGFß is a paradigm morphogen during embryonic development, and a pleiotropic cytokine of adult tissues. In recent years, we have been interested in dissecting what are the cellular determinants that limit the repertoire of gene-expression programs that TGFß can induce in a given cellular context. Our work has revealed the importance of p53 family members in directing TGFß gene responses in embryonic development and adult epithelial cells (Cordenonsi et al., Cell 2003; Science 2007). We have also identified, and genetically validated, a new set of enzymes regulating intensity and duration of TGFß signaling by causing regulative ubiquitination and de-ubiquitination of Smads (Dupont et al., Cell 2005; Cell 2009; Inui et al., Nat Cell Biol 2010; Morsut et al., Development 2010), and contributed to the concept that TGFß signaling is under negative control by secreted antagonists and miRNAs (Zacchigna et al., Cell 2006; Martello et al., Nature 2007).
|
|
|
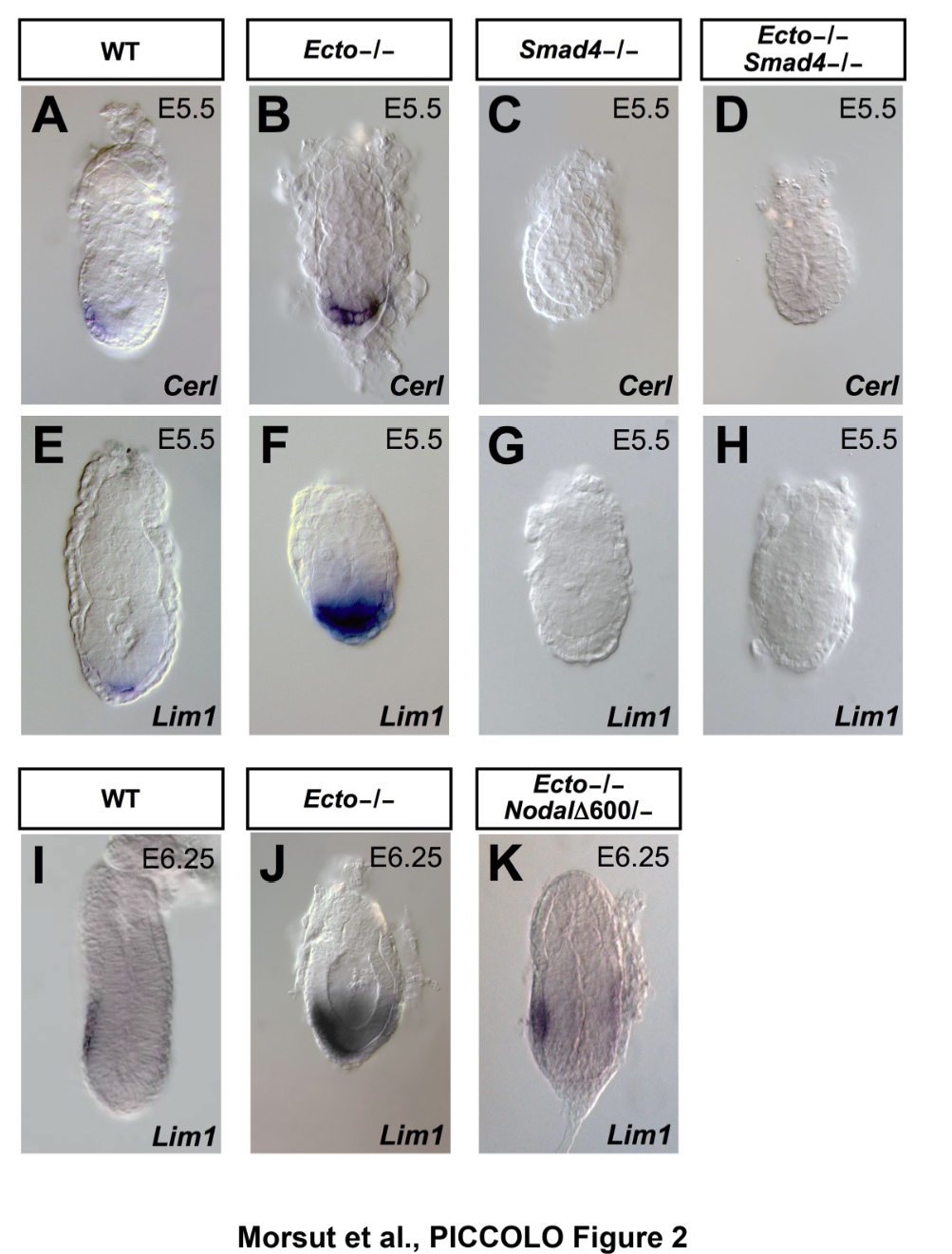
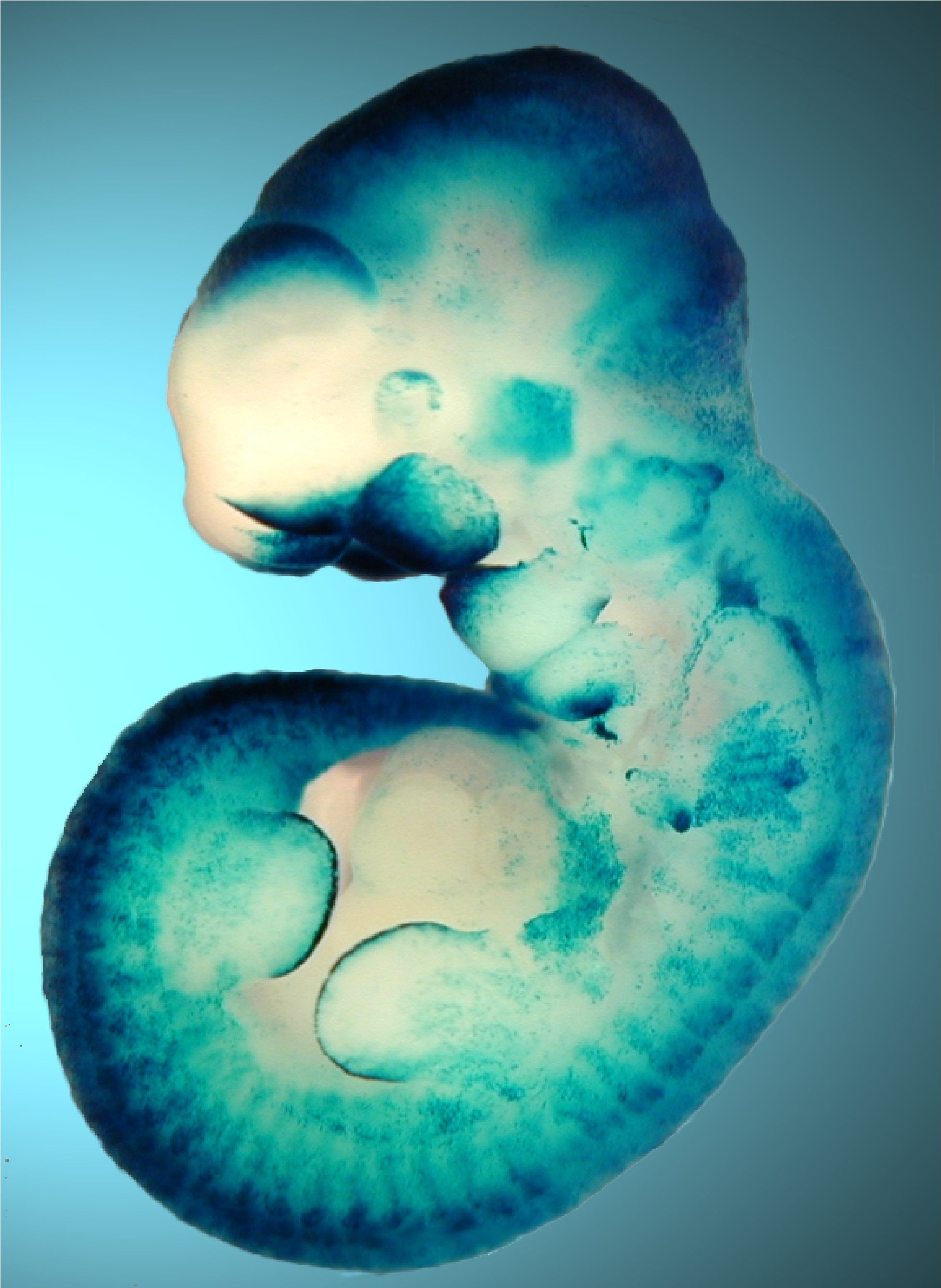
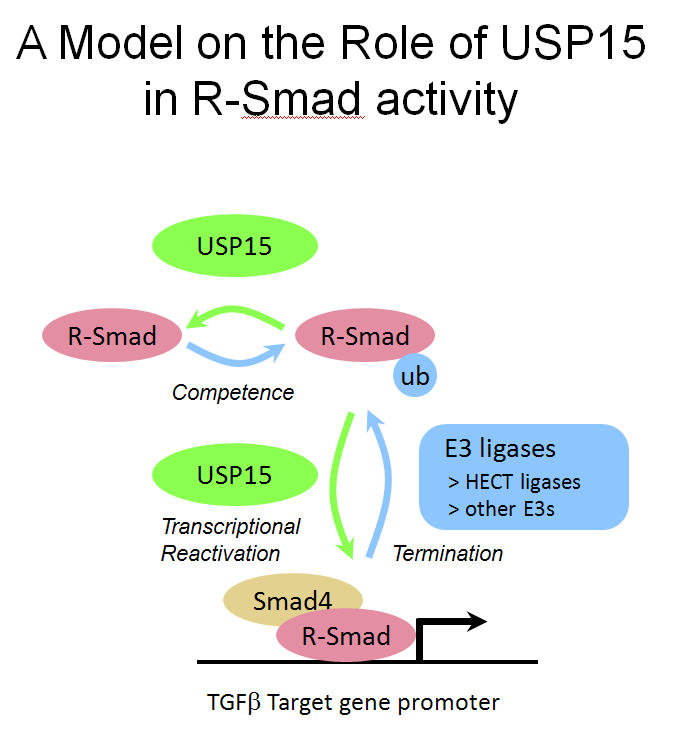
(Left) The pictures show mouse embryos at very early stages of development, stained for Smad target genes. Absence of the Smad inhibitor Ectodermin/Tif1g/TRIM33, enhanced endogenous TGFb signaling; mutant mice are genetically rescued by reducing the dosage of TGFb (Nodal) ligand (Morsut et al., 2010; Dupont et al., 2009). (Right) The Wnt-reporter BAT-gal mouse (Maretto et al., 2003). (Bottom) A model for R-Smad regulation by the ubiquitin system (Inui et al., 2011). |
|
|
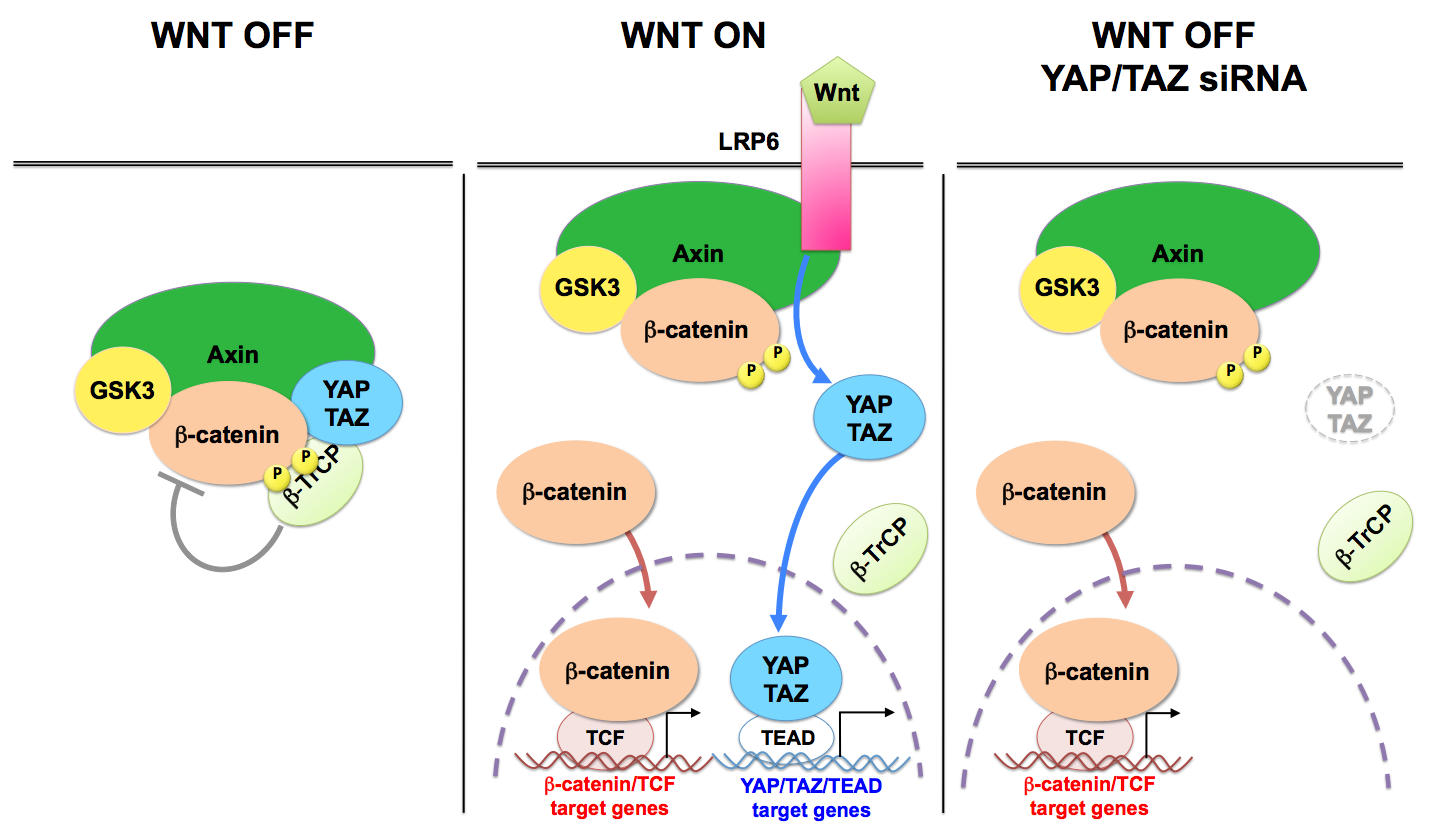
Constitutive activation of the Wnt pathway is a main oncogenic input for several tissues. Wnts are secreted cytokines that interact with membrane receptors, Frizzled and LRP5/6, leading to the inactivation of a cytoplasmic protein complex, named the b-catenin destruction complex. Within this complex, two central scaffold proteins, Axin1/2 and adenomatous polyposis coli (APC), recruit the kinase glycogen synthase kinase-3 (GSK3) to phosphorylate b-catenin promoting its degradation. |
We recently found that, in absence of Wnt stimulation, cytoplasmic YAP/TAZ contribute to the function of the destruction complex (Azzolin et al., 2014) (see Figure). YAP/TAZ bind directly Axin, APC and b-catenin, causing their sequestration in the complex, and also causing TAZ to interact with b-TrCP, leading to its degradation (Azzolin et al., 2014; Azzolin et al., 2012). Therefore, Wnt stimulation, depletion of Axin1/2 or of APC release YAP/TAZ from such cytoplasmic sites and promote YAP/TAZ nuclear accumulation and target genes transcription (Azzolin et al., 2014; Azzolin et al., 2012; Byun et al., 2014). YAP/TAZ are upregulated and nuclear after APC loss in the intestine (Azzolin et al., 2014; Cai et al., 2015; Camargo et al., 2007; Gregorieff et al., 2015). Functionally, YAP/TAZ are required downstream to APC mutations for uncontrolled proliferation of the intestinal epithelium and intestinal tumorigenesis (Azzolin et al., 2014; Cai et al., 2015; Gregorieff et al., 2015). In CRC cells bearing inactivating APC mutations, b-catenin and TAZ cooperate to promote cell growth (Azzolin et al., 2012).
Nuclear localization of YAP/TAZ is by itself an event that depletes the destruction complex favoring b-catenin nuclear localization; for example nuclear translocation of YAP/TAZ induced by strong mechanical stimuli - such as those produced by inflammation, fibrosis, or stretching epithelial monolayers - has been recently shown to be sufficient to be followed by b-catenin activation even without overt presence of Wnt ligands (Nowell et al., 2016). Thus, the mechanical control of YAP/TAZ can feedback into the same pathways that control tissue patterning, providing a mean for the cell's structural features to attain a high-order control over growth factor signaling.